

By merging stem cell and organ-on-a-chip technologies, researchers have grown functioning, yet diseased, human heart tissue. The technology is already put to use as a testing platform for drugs that might treat inherited heart diseases.
Barth syndrome is a rare, untreatable cardiac disorder that’s caused by the mutation of a single gene, TAZ, which encodes for Tafazzin. The X-linked disorder appears mostly in boys and comes with a range of symptoms affecting the function of heart and skeletal muscle.
A team led by Harvard Stem Cell Institute’s Kevin Kit Parker and William Pu of Children's Hospital Boston took skin cells from two Barth syndrome patients and turned them into stem cells that carrying the TAZ mutations. They grew the stem cells on chips that were lined with human extracellular matrix proteins, the biochemical support. By mimicking their natural environment, the team tricked the cells into joining together as if they were forming a diseased human heart.
And it worked: The engineered diseased tissue contracted weakly, like the heart muscle of a patient. In this video, you can see the difference in contractile strength of engineered cardiac tissue derived from a healthy patient and a Barth syndrome patient. Healthy tissue generates a more lot more stress with each contraction, while diseased tissue might not move at all.
Using genome editing, the team mutated TAZ in normal cells to confirm that the mutation causes the weak contraction. Then, by delivering wildtype copies of the TAZ gene product to the diseased tissue, the team corrected the contractile defect. Behold, the first tissue-based model of a genetic heart disease, corrected!
The team also discovered that the TAZ mutation disrupts the activity of mitochondria, the energy-making organelles. But it didn’t affect the overall energy supply of the cells; the team may have stumbled upon a link between mitochondrial function and the ability of a heart cell to build itself in a way that lets it contract. “The TAZ mutation makes Barth syndrome cells produce an excess amount of reactive oxygen species, or ROS -- a normal byproduct of cellular metabolism released by mitochondria,” Pu explains in a news release. Quenching the excessive ROS production restores contractile function.
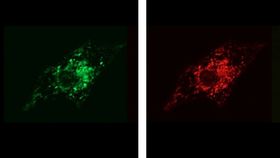
This series of images shows how inserting the modified genetic material into diseased tissue causes the cells to produce functioning versions of the TAZ protein (green), which correctly localize in the mitochondria (red). The images are merged to create the last picture, demonstrating this localization (yellow).

“You don’t really understand the meaning of a single cell’s genetic mutation until you build a huge chunk of organ and see how it functions or doesn’t function,” Parker says. “In the case of the cells grown out of patients with Barth syndrome, we saw much weaker contractions and irregular tissue assembly. Being able to model the disease from a single cell all the way up to heart tissue, I think that’s a big advance.”
The work was published in Nature Medicine this week.
Image: Gang Wang and William Pu, Children’s Hospital Boston