


Establishing the shapes of proteins is a major challenge for science, but turning the problem into a game is speeding up the hunt for solutions. Players of Foldit, where online teams compete to work out likely shapes, are proving better at it than scientists or computer programs designed for the purpose. The beaten researchers may have had their pride dented, but were compensated with solutions to a problem they struggled with themselves.
The rest of this article is behind a paywall. Please sign in or subscribe to access the full content.For years now scientists have been using idle processing power to analyze tricky data, most famously in SETI@home, where data from radio telescopes is distributed to three million computers to search for possible alien signals.
Confronted with a problem computers still are not all that good at, Dr Scott Horrowitz of the University of Michigan turned to the idle processing power in gamers' heads. He recruited 469 Foldit players, two professional crystallographers – for whom solving protein shapes is their daily work – and 61 undergraduates at his university. Two computer programs designed for this purpose were given the same problem to keep the humans on their toes.
All were given electron density maps and sequences for a yeast protein, YPL067C, and challenged to work out the shape. The crystallographers and students worked independently, but the gamers formed themselves into teams.
The results were published in Nature Communications. Old proverbs about many heads being better than one were proven true as the Foldit players produced solutions that provide a better fit with what we know than the more experienced individuals. One Foldit player provided several particularly important components of the final shape, but gained valuable assistance from other team members' refinements.

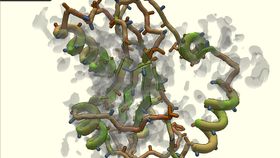
Stages in Foldit player's development of a model for the protein YPL067C. Horowitz et al/Nature
“I've seen how much players learn about proteins from playing this game," said Horowitz. "We spend weeks and weeks trying to jam this into students' brains and Foldit players learn it naturally because it's fun."
The Foldit players used a different approach to the crystallographers and students. It is possible each could learn from the other to do better still. The computer algorithms performed substantially worse than either set of humans.
Proteins contain so many atoms that there are enormous numbers of ways they could be structured, and working out the exact structure can be essential to finding drugs that bind to them. Solving protein folds is such a challenging problem Randall Munroe of XKCD wrote, “Someone may one day find a harder one”. It has been estimated that 85 percent of the structures molecular biologists use as approximations of protein shapes contain discernible errors, undermining further research.
Techniques such as X-ray diffraction have enabled us to gain some idea of protein shapes, but the problem is so challenging that prominent British Chemist George Sheldrick said; “Macromolecular refinement against high-resolution data is never finished, only abandoned.” By capturing the enthusiasm of the gaming community Horowitz may have ensured that the problems are never truly abandoned as unsolvable either.


